Heme proteins and Protein-based Radicals
Centre National de la Recherche Scientifique (CNRS)
Directeur de Recherche CNRS
Permanent Research position,
attached to UMR 7281 since Sept 2014.
 https://orcid.org/0000-0002-4275-0980
https://orcid.org/0000-0002-4275-0980
Education:
1986: MSc in Physics, University of Rosario, Department of Physics, Argentina.
1991: PhD in Physics, University of La Plata, Department of Physics. Argentina.
1999 : Qualification aux fonctions de Professeur des Universités. Conseil National des Universités. Paris, France.
2007 : Habilitation à Diriger des Recherches (HDR). Université Paris-Sud XI. Département de Chimie. Orsay, France.
Previous Appointments:
* 2012-2014 : Directeur de Recherche CNRS. Permanent CNRS position attached to UMR 8221, CEA/CNRS/Paris-Sud University. Saclay, France.
(Note : same Research Unit as previous appointment(s), just changed name).
* 2001-2012 : Chargé de Recherche CNRS. Permanent CNRS position attached to URA 2096, CNRS/CEA. Saclay, France.
* 2000-2001 : Chercheur Associé CNRS. Temporary CNRS research position from the URA 2096, CNRS/CEA. Saclay, France.
* 1998-2000 : Research Fellow CEA-Saclay. Temporary research position from the URA 2096, CNRS/CEA. Saclay, France.
(Multifrequency EPR and Resonance Raman spectroscopies on catalytic intermediates in heme catalases and peroxidases, Tyr● and Trp● intermediates).
* 1997-1999 : Chargé de Recherche CONICET. Permanent position, Argentinian Research Council (CONICET), attached to the IFLYSIB Research Institute, La Plata, Argentina.
(Note: I resigned my position in 1999 to apply for a permanent CNRS position in France).
* 1993-1996 : postdoc. French Atomic Energy Commission, CEA-Saclay and CEA-Grenoble. France.
– URA 2096, CNRS/CEA. Saclay (Pulsed EPR spectroscopy on catalytic intermediates in manganese catalases and Resonance Raman spectroscopy on reactive intermediates in Photosynthetic reaction centers).
-Service de Chimie Inorganique et Biologie, CEA-Grenoble (multifrequency EPR spectroscopy on catalytic intermediates in heme catalases and Tyr● intermediates).
* 1991-1992 : postdoc, Biological Research Center, Institute of Biophysics. Szeged, Hungary. (supervisor Dr Laszlo I. Hórvath; Spin label EPR spectroscopy on lipid-protein interaction in Photosynthetic reaction centers).
Research:
Our Research focuses on understanding the molecular aspects of the various biologically-competent catalytic pathways in heme-containing proteins. We are particularly interested on the identification and characterization of redox-active Trps and Tyrs, having a concerted role with the heme cofactor in enzyme catalysis. The [Fe(IV)=O Trp●] and/or [Fe(IV)=O Tyr●] intermediates are key players in specific oxidation reactions, involving long-range ET pathways in the catalytic cycles of certain heme enzymes. Formation of the Trp/Tyr-based high-valent intermediates results from the one-electron intramolecular electron transfer (ET) subsequent to the formation of [Fe(IV)=O Por●+], the primary high-valent intermediate in heme enzymes catalysis. Such protein-based radical intermediates provide with alternative reactivity that crucially expands the catalytic reactions of heme enzymes. Our goal is to understand Nature’s strategies to selectively fine tune the reactivity of Trp and Tyr as oxidation sites and/or electron relays in enzyme catalysis. Such fundamental and detailed knowledge is necessary to better exploit natural catalysts, and ultimately to design and develop bioinspired artificial mini-metalloproteins, as tunable catalysts to be used in chemical and pharmaceutical industrial applications. Accordingly, our work includes both natural and synthetic heme catalysts.
Role of [Fe(IV)=O Trp●] in heme catalysis:
Catalase-peroxidases, known as KatGs, are bifunctional heme enzymes which arose via gene duplication of an ancestral hydroperoxidase. This family of enzymes has been central to our work (collaborations with Prof. Peter C. Loewen, Department of Micrbiology, University of Manitoba, Winnipeg, Canada, and Dr Wladimir Sougakof, Research Center for Immunology and Infectious Deseases (CIMI), Paris Sorbone University, France) and it has provided us with a naturally-occurring, and well-defined, diversity in ET pathways (3) in selected members of the KatG family from both pathogenic (M. tuberculosis and B. pseudomallei) and non-pathogenic bacteria (Synechocystis, Synechococcus, E. coli) (7, 8, 12, 14, 15, 16, 17). These bifunctional enzymes are involved in the cellular response and regulation of oxidative stress (catalase-like enzymatic reaction). Their physiological role as catalases is less well understood. In addition, the KatG from Mycobacterium tuberculosis is responsible for the activation of isoniazid (isonicotinic acid hydrazide, INH), the front-line prodrug used in the treatment against tuberculosis infection. We have shown that such INH prodrug activation occurs via a [Fe(IV)=O Trp●] intermediate (14), and not through the heme-edge reaction (see Figure 1, panel B). One other singularity of KatGs is the unprecedented post-translational modification, a Trp-Tyr-Met adduct (see Figure below), on the distal heme side which together with a pH-dependent mobile Arg residue (15) are essential in the disproportionation of hydrogen peroxide, thus differing substantially from the reaction mechanism in monofunctional heme catalases (3).
- pseudomallei and M. tuberculosis KatGs:

Figure 1: Panel A (see Ref. #2): Structure of the N-terminal domain of B. pseudomallei KatG (BpKatG) with isoniazid (INH) bound (PDB reference 3N3N). The cross-sectional view, perpendicular to the main access channel to the heme active site, shows the heme environment. The three radical sites shown in green (Trp330, Trp139, Trp153) correspond to the [Fe(IV)=OTrp●] intermediates, discerned by multifrequency (9-285 GHz) EPR spectroscopy on single, double, and triple Trp variants in BpKatG (11). The Trp residues in blue (Trp111, Trp94, Trp95) were characterized as electron relays, that together with the structural water molecules (shown in red) assist the long-range ET between Trp153 and/or Trp139 to the heme. The paths are indicated by blue and white dashed lines. The long distance (24.6Å) between the INH substrate and the heme precludes single-step tunneling phenomena to occur at biologically relevant rates. Accordingly, we have shown that the [Fe(IV)=O Trp●139] is the actual reactive intermediate for the INH prodrug activation, and not the [Fe(IV)=O Por●+](12, 14). Hence, multistep tunneling is likely to occur for the oxidation of INH, with distances of 8.1 Å from INH to Trp202, 4.5 Å from Trp202 to Trp139, and 9.7 Å fromTrp139 to Trp111.
Figure 1: Panel B (see Ref.#2 below): Surface view of BpKatG with isoniazid (INH) bound in a lateral funnel-shaped channel. The surface is rendered with partial transparency to reveal the buried heme site on the opposite side of the protein, with a distance of ca. 25 Å between the heme iron and the INH substrate. Taken together, the findings that the prodrug does not bind close to the heme and that the [Fe(IV)=O Trp●] is the reactive intermediate for INH oxidation, challenges the conventional view of KatG behaving like a typical peroxidase. This is crucial for drug design, that so far has considered a heme-edge reaction.
Our early detailed multifrequency EPR/stopped-flow kinetics/site-directed mutagenesis studies on the non-pathogenic Synechocystis KatG (SynKatG) (16) showed that there is a unique Trp site for the formation of the [Fe(IV)=O Trp106●] intermediate (14), in sharp contrast to the three Trp sites in BpKatG (see below).

An important point is that such a unique Trp106 site in SynKatG (corresponding to Trp95 in BpKatG numbering, see Figure 3) plays the role of electron relay in BpKatG.

Consistently, we have also shown in the triple-Trp variant of BpKatG, that removes the three naturally-occurring Trp sites in the B. pseudomallei enzyme (i.e. Trp139, Trp153, and Trp330; Fig. 3), stabilizes Trp95● (11), the equivalent Trp position of the unique radical site in SynKatG. We have also shown that the [Fe(IV)=O Trp106●]intermediate in SynKatG is not the reactive intermediate for INH (9), thus reflecting the differences in substrate accessibility to the heme site and kinetics that have been overlooked in previous studies by other groups.
Such a naturally-occurring selectivity in radical sites for the catalytic [Fe(IV)=O Trp●] intermediate within the same family of KatG enzymes provides us with ample opportunities and challenges to understand better and to harness the physicochemical properties and factors modulating the specific stabilization and reactivity of Trp radicals and the high-valent heme iron-radical intermediates in other natural heme and non-heme enzymes. For example, the unusual high oxidizing potential of the [Fe(IV)=O Trp●] intermediate in lignin peroxidase that is the reactive species for veratryl alcohol (substrate) oxidation (E0 = 1.36V), the later being the mediator for the radical-mediated reaction to depolymerize lignin (11). We have shown that the stability of the Trp radical site can be modulated by the microenvironment of the Trp (11). We also suceeded in engineering the Trp radical site and its reactivity as [Fe(IV)=O Trp●] intermediate in a different peroxidase, C. cinereus (10). Also, differences in electron transfer reactions observed between yeast CcP and Lehismania major peroxidase are currently studied (collaboration with Prof. Thomas Poulos, University of California, Irvine, USA).
We are also exploring the complementary reactivity of lignolitic fungal oxidoreductases, having heme or copper active site(s) (collaboration wih Prof. Craig B. Faulds, INRA/Aix-Marseille University, UMR 1163 « Biodiversité et Biotechnologie Fongique », Marseille, France).
Role of [Fe(IV)=O Tyr●] in heme catalysis:
We succeeded in clarifying the long-lasting controversy (16) about Tyr● (i.e. « radicals all over ») in cytochrome c peroxidase (CcP), by carrying out a systematic investigation of the location of the Tyr radical(s) and the putative role in substrate oxidations. The use of multifrequency (9-285 GHz) EPR spectroscopy combined with carefully designed multiple-site Trp/Tyr mutations (collaboration with Prof. Yi Lu, University of Illinois at Urbana-Champaign, Urbana, USA) was crucial in order to identify Tyr71 and Tyr236 as those contributing primarily to the EPR Tyr• signal in CcP (6). Moreover, we monitored the specific reactivity of the high valent intermediates by using EPR spectroscopy, and showed that [Fe(IV)=O Tyr71●] is the reactive intermediate in the oxidation of guaiacol substrate, binding at a site remote from the heme, with Trp51 (on the distal heme site) being involved in the long-range ET pathway between Tyr71 and the heme (6). The strategy used for identifying the elusive Tyr radical sites in CcP, in particular the design of mutations that destabilize formation of the Tyr● without actual substitution of the Tyr site, may be generalized to other heme enzymes containing a high number of Tyr and Trp residues and for which Tyr (or Trp) radicals have been proposed to be involved in their peroxidase or peroxidase-like reaction, such as Chloride dismutases, Dye-decoloring peroxidases, peroxygenases, and nitrophorins.
Nitrophorins : In order to address the question on the role of the ferric resting state of the heme active site in nitrophorins, that is unusual for an NO-carrier/binding enzyme, we studied the reactive intermediates and putative alternative reactivities of R. prolixus nitrophorin (isoform 2) (9), in collaboration with Prof. F. Ann Walker, University of Arizona, Tucson, USA. A comprehensive study using EPR and stopped-flow electronic absorption spectroscopies (9) demonstrated an unprecedented reaction of the high-valent intermediate with norepinephrine at physiological pH, (Figure 4) that is indicative of an alternative strategy to neutralize the effect of such a molecule when released into the blood stream as a consequence of the insect’s bite. Hence, nitrophorin behaves as NO carrier when exposed to the acidic pH of the insect saliva. If exposed to the higher pH of the tissues and the capillaries, NP2 in its ferric state will either capture histamine or, in the usual presence of hydrogen peroxide, it will efficiently inactivate norepinephrine through a peroxidase-like reaction (9).
Similarly, we could address the long-lasting controversy about protein-based radicals observed in lactoperoxidase (LPO) (13). A particular emphasis was given to determine the chemical nature of the protein-based radical intermediates that have been theoretically proposed or indirectly detected in previous studies. Moreover, we chose a mammalian heme peroxidase since they show different oxidizing capabilities as well as a high number of Try and Tyr, as the case of bifunctional KatGs. The radicals were discerned by the advantageous resolution of their g-tensors, resolved with higher field/high frequency (10 T/285 GHz) EPR measurements. In the substrate-free reaction of lactoperoxidase with hydrogen peroxide, a Tyr● and a Trp● were formed subsequent to the [Fe(IV)=O Por●+] intermediate. A combination of EPR and stopped-flow measurements (Figure 5) was used to characterize the reaction of the LPO enzyme with two sets of substrates, small and bulky ones and expected to be oxidized by either heme-edge reaction (o-dianisidine, o-anisidine, benzohydroxamic acid) or a long-range electron transfer route (ABTS and mitoxantrone).
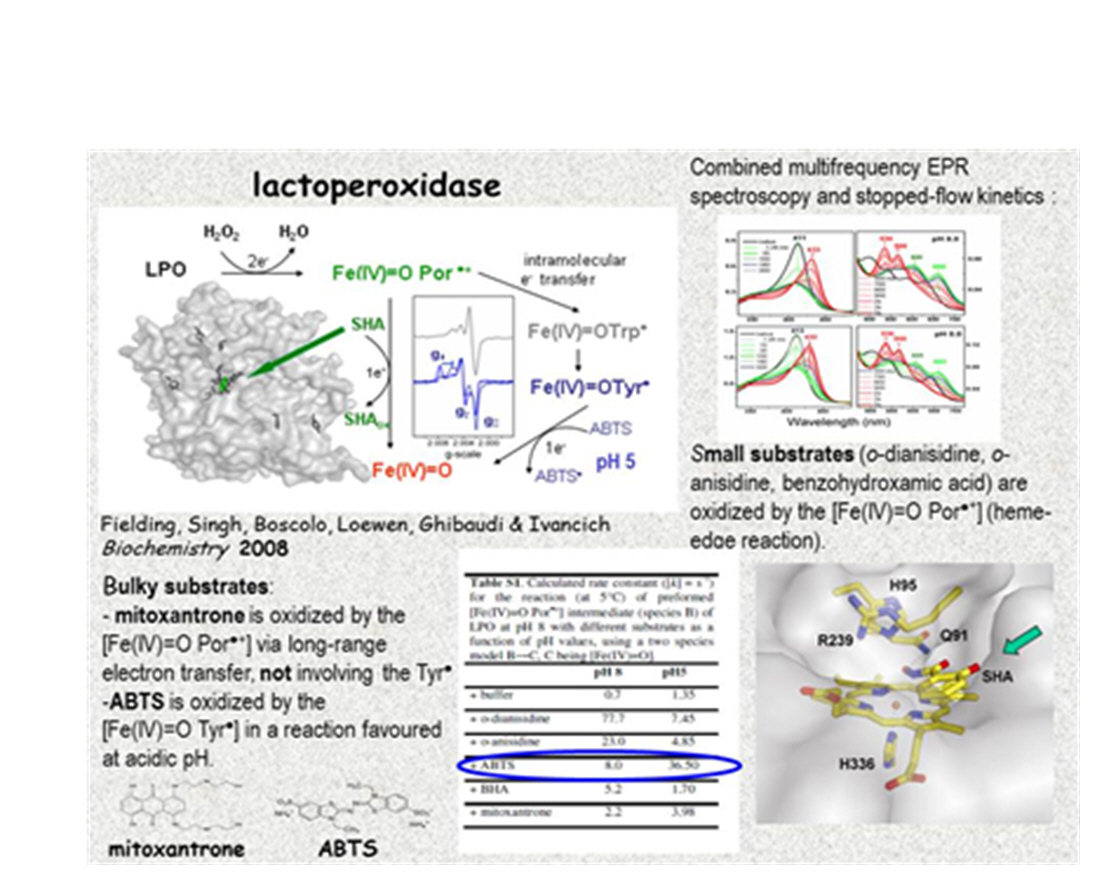
The observed pH-dependent competition between oxidation of mitoxantrone by the [Fe(IV)=O Por●+] intermediate, via long-range electron transfer and the formation of the protein radical intermediates indicated kinetic relevance of the latter. Moreover, the [Fe(IV)=O Tyr●] was concluded to be the oxidizing species for ABTS in a reaction favoured at acidic pHs (13). Current complementary studies using Resonance Raman spectroscopy (in collaboration with Prof. James R. Kincaid, Department of Chemistry, Marquette University, Milwaukee, USA) and molecular dynamics and Monte Carlo sampling technique (PELE) to model ABTS binding (in collaboration with Prof. Victor Guallar, Barcelona Supercomputing Centre, Barcelona, Spain) will demostrate the requirement of all three spectroscopies (EPR, RR, and stopped-flow electronic absorption) plus the computational approach to assess Nature’s strategies expanding the classical heme-edge chemistry via the [Fe(IV)=O Tyr●] intermediate and selected Tyr● facilitating long-range electron transfer.
Last but not least, we have revisited the enzymatic reaction of catalases in the perspective of an alternative function and inspired on the role of protein-based radical intermediates in peroxidases and KatGs. Catalases are responsible for the regulation of hydrogen peroxide (H2O2) content in the cell to avoid the toxic effects in oxidative stress conditions. Catalases catalyze the disproportionation of H2O2 to molecular oxygen and water at a very high rate, and without being inactivated in the presence of very high levels of H2O2 (3). Such a catalytic reaction happens through the [Fe(IV)=O Por●+] intermediate, with the heme site being deeply buried in the protein and with a restrictive access channel (Figure 6).
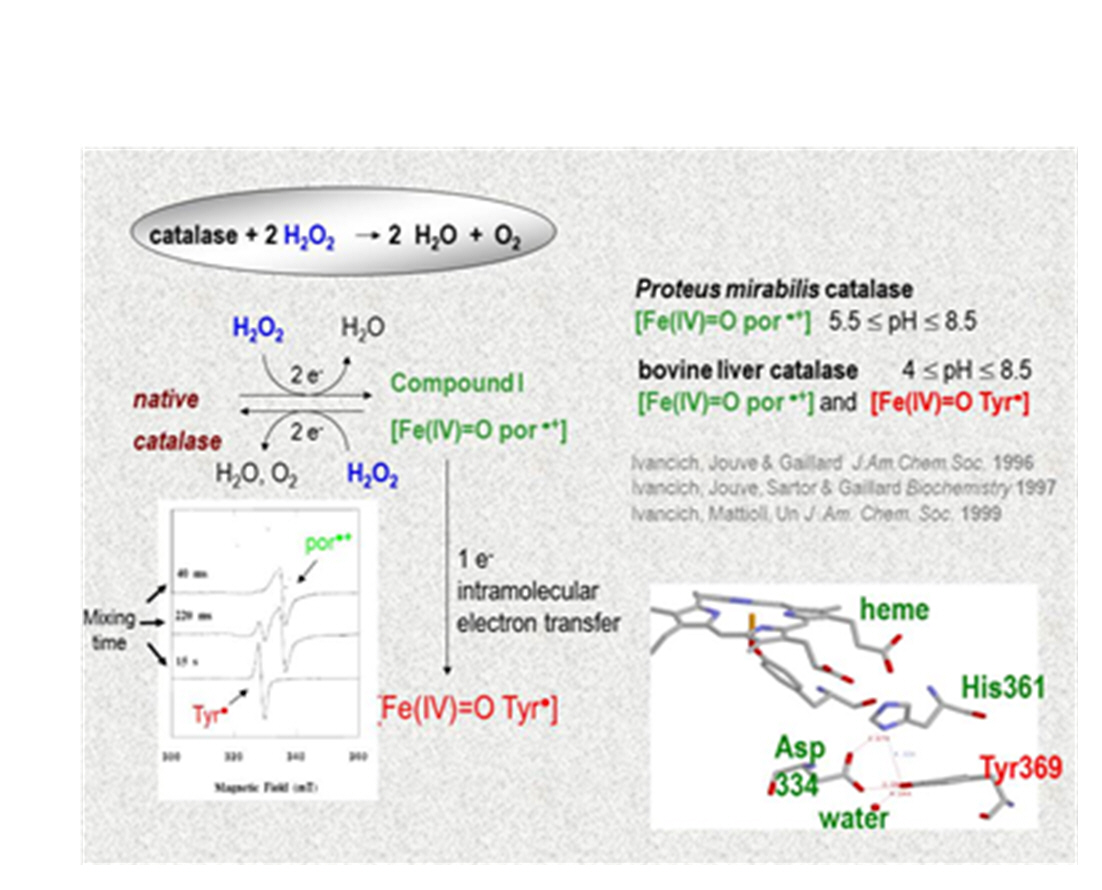
Yet, our early studies using a combined approach of 9-285 GHz EPR spectroscopy, rapid-mix freeze-quench technique and Resonance Raman spectroscopy on bovine liver catalase showed the catalytically competent formation of an unprecedented [Fe(IV)=O Tyr●] intermediate (19, 20, 21). At the time, bovine liver catalase was considered as a “one off” case. Yet, our more recent findings (collaboration with Prof. Peter C. Loewen, University of Manitoba, Winnipeg, Canada) on the pH-dependent formation of [Fe(IV)=O Tyr●] and [Fe(IV)=O Trp●] intermediates in the catalase from Helicobacter pylori, a human pathogenic bacteria and the unprecedented structural/functional features of a catalase from Bacillus pumilus (5), a soil bacteria, showing access and binding of phenolic substrates to the heme active site in a peroxidase-like fashion and including protein-based radical intermediates, strongly suggest that ’monofunctional’ catalases have also evolved to use alternative strategies for distinct biological functions (3), similar to peroxidases and KatGs.
Artificial mini-heme proteins: Our recent efforts are oriented to the combined investigation of KatGs as natural catalysts together with artificial enzymes, exploiting the emerging field of de novo metalloprotein design (the latter in collaboration with Prof. Vincent L. Pecoraro, Department of Chemistry, University of Michigan, Ann Arbor, USA). as well as using iron porphyrin complexes as cofactors (2) (in collaboration with Prof. Abhishek Dey, Indian Association for the Cultivation of Science, Kolkata, India).
The protein environment plays a crucial role in tuning the different reactivities of the heme cofactor. Differences in the amino acids being the ligand to the iron and the close-by H-bonds and electrostatic interactions convert a lignin peroxidase to a cyt P450 monooxygenase. Man-made synthetic heme proteins serve to scrutinize these natural tuning strategies and to produce efficient green catalysts for pharmaceutical and industrial applications. We have synthetized peptides that self‐assemble as α‐helical coiled coils, as shown in Figure 7, to provide a switchable environment to the heme cofactor (1). UV-vis and Electron Paramagnetic Resonance spectroscopies were used to identify the differences in heme coordination as a function of pH.
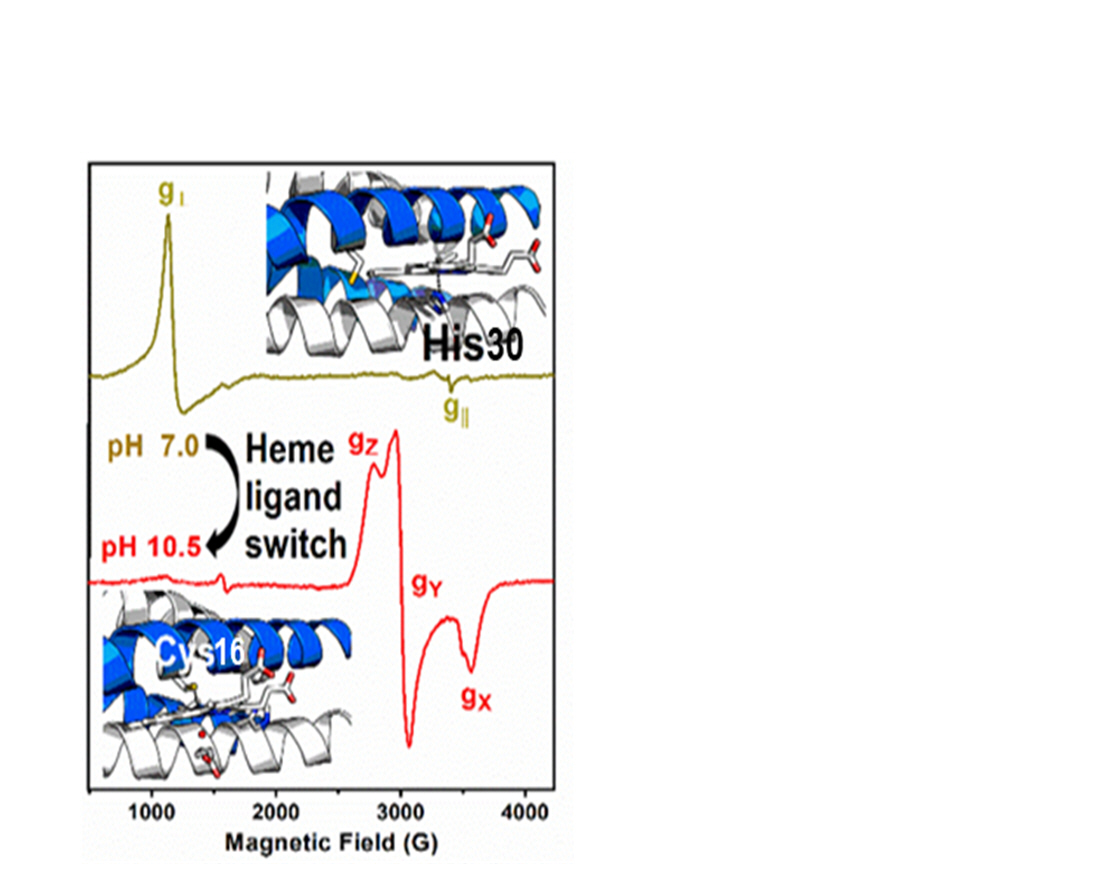
We were able to assess the preference of the heme cofactor when having the opportunity to bind either to histidine or to cysteine, thus uncovering a previously undescribed pH-dependent switch in the heme binding mode within a single synthetic protein (1). The resulting miniature protein, that folds as a dimer of antiparallel two-stranded coiled coils upon heme complexation, can catalyze O2 reduction (as cyt P450 monooxygenases) as well as substrate oxidation (as peroxidases). Accordingly, the designed α‐helical scaffold opens exciting opportunities to explore heme enzyme chemistry.
Representative Publications:
(1) Koebke, K.J, Kühl, T., Lojou, E., Demeler, B., Schoepp-Cothenet, B., Iranzo, O., Pecoraro, V.L.*, Ivancich, A.* (2021) Angew. Chem. Int. Ed. 60 (8), 3974-3978.
(2) “A designed second-sphere hydrogen-bond interaction that critically influences the O–O bond activation for heterolytic cleavage in ferric iron–porphyrin complexes » Bhunia, S., Rana, A., Ghosh Dey, S., Ivancich, A.*, Dey, A.* (2020) Chem. Sci. 11, 2681-2695. https://doi.org/10.1039/C9SC04388H
(3) “Electron Transfer in Catalases and Catalase-Peroxidases” Ivancich, A.*, Loewen P.C. (2018) in: Encyclopedia of Biophysics, European Biophysical Societies’ Association (EBSA) 2018 (Roberts G., Watts A., Eds). Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-35943-9_51-1
(4) “An alternative reaction for heme degradation catalyzed by the Escherichia coli O157:H7 ChuS protein : Release of hematinic acid, tripyrrole and Fe(III)” Y. H. Ouellet, C. T. Ndiaye, Gagné, S., Sebilo, A., Suits, M. D. L., Jubinville, J., Jia, Z., Ivancich, A.*, Couture, M.* (2016) J. Inorg. Biochem. 154, 103-113.
(5) “Unprecedented access of phenolic substrates to the heme active site of a catalase : Substrate binding and peroxidase-like reactivity of Bacillus pumilus catalase monitored by X-ray crystallography and EPR spectroscopy” Loewen, P.C., Villanueva, J., Switala, J., Donald, L.J., Ivancich, A. (2015) Proteins : Structure, Function, and Bioinformatics 83, 853-866.
(6) “Identifying the elusive sites of tyrosyl radicals in cytochrome c peroxidase : Implications for oxidation of substrates bound at a site remote from the heme” Miner, K. D., Pfister, T. D., Hosseinzadeh, P., Karaduman, N., Donald, L.J., Loewen, P.C., Lu, Y., Ivancich, A.* (2014) Biochemistry 53, 3781-3789.
(7) “Spectroscopic and kinetic investigation of the reactions of peroxyacetic acid with Burkholderia pseudomallei catalase−peroxidase, KatG” Ivancich, A., Donald, L.J., Villanueva, J., Wiseman, B., Fita, I., Loewen, P.C. (2013) Biochemistry 52, 7271−7282.
(8) “Structure-function relationships in heme peroxidases: New insights from electronic absorption, resonance Raman and multifrequency Electron Paramagnetic Resonance spectroscopies” Smulevich, G., Feis, A., Howes, B.D., Ivancich, A. (2010) in Handbook of Porphyrin Science (Kadish, K. M., Smith, K. M., Guilard, R. Eds), vol. 6, chapter 31, pp 367-453, World Scientific, Hackensack, NJ.
(9) “Unprecedented peroxidase-like activity of Rhodnius prolixus Nitrophorin 2 : Identification of the [FeIV=O Por ●]+ and [FeIV=O Por](Tyr38●) intermediates and their role(s) in substrate oxidation” Singh, R., Berry, R.E., Yang, F., Zhang, H., Walker, F.A., Ivancich, A.* (2010) Biochemistry 49, 8857–8872.
(10) “The reaction of Synechocystis PCC6803 catalase-peroxidase (KatG) with isoniazid investigated by multifrequency (9-285 GHz) EPR spectroscopy” Colin, J., Jakopitsch, C., Obinger, C., Ivancich, A.* (2010) Appl. Magn. Reson. 37, 267-277.
(11) “Spectroscopic evidence for an engineered, catalytically active Trp radical that creates the unique reactivity of lignin peroxidase” Smith, A.T., Doyle, W.A., Dorlet, P., Ivancich, A.* (2009) Proc. Natl. Acad. Sci. USA 106, 16084-16089.
(12) “Distinct role of specific tryptophans in facilitating electron transfer or as [Fe(IV)=O Trp●] intermediates in the peroxidase reaction of Bulkholderia pseudomallei catalase-peroxidase : A multifrequency EPR spectroscopy investigation” Colin, J., Wiseman, B., Switala, J., Loewen, P.C, Ivancich, A.* (2009) J. Am. Chem. Soc. 131, 8557-8563.
(13) “Intramolecular electron transfer vs. substrate oxidation in lactoperoxidase : Investigation of the radical intermediates by stopped-flow absorption spectrophotometry and EPR spectroscopy” Fielding, A.J. , Singh, R., Boscolo, B., Loewen, P.C., Ghibaudi, E.M., Ivancich, A.* (2008) Biochemistry 47, 9781−9792.
(14) « Two [Fe(IV)=O Trp●] intermediates in M. tuberculosis catalase-peroxidase discriminated by Multifrenquency (9−285 GHz) EPR Spectroscopy : Reactivity towards isoniazid » Singh, R., Switala, J., Loewen, P.C., Ivancich, A.* (2007) J. Am. Chem. Soc. 129, 15954-159563.
(15) « Identification of Trp106 as the tryptophanyl radical intermediate in Synechocystis PCC6803 catalase-peroxidase by multifrequency Electron Paramagnetic Resonance spectroscopy » Jakopitsch, C., Obinger, C., Un, S., Ivancich, A.* (2006) J. Inorg. Biochem. 100, 1091-1099.
(16) » A molecular switch and electronic circuit modulate activity in catalase-peroxidases » Carpena, X., Wiseman, B., Deemagarn, T., Singh, R., Switala, J., Ivancich, A., Fita, I., Loewen, P.C (2005) EMBO Reports 6, 1156-1162.
(17) « Protein-based radical intermediates in the catalase-peroxidase of Synechocystis PCC6803 : A multifrequency EPR investigation of wild type and variants on the environment of the heme active site” Ivancich, A., Jakopistch, C., Auer, M., Un, S., Obinger, C. (2003) J. Am. Chem. Soc. 125, 14093-14102.
(18) « Multifrequency High-Field EPR study of the Tryptophanyl and Tyrosyl radical intermediates of wild type and the W191G mutant of cytochrome c peroxidase ». Ivancich, A.*, Dorlet, P., Goodin, D.B., Un, S. (2001) J. Am. Chem. Soc. 123, 5050-5058.
(19) « The effect of protein microenvironment on Tyrosyl radicals. A High Field (285 GHZ) EPR, Resonance Raman and Hybrid Density Functional study ». Ivancich, A., Mattioli, T.A., Un, S. (1999) J. Am. Chem. Soc. 121, 5743-5753.
(20) « EPR Investigation of compound I in Proteus mirabilis and bovine liver catalases : Formation of Porphyrin and Tyrosyl radical Iintermediates ». Ivancich, A., Jouve, H.M., Sartor, B., Gaillard, J. (1997) Biochemistry 36, 9356-9364.
(21) « EPR evidence for a Tyrosyl radical intermediate in bovine liver catalase » Ivancich, A., Jouve, H.M., Gaillard, J. (1996) J. Am. Chem. Soc. 118, 12852-12853.