A tutorial on Protein Film Voltammetry (PFV)

Enzymes have a business: they catalyse a reaction. And for more than one hundred years, studying how enzymes work has involved studying the rates of the reactions they catalyse.
For mechanistic studies of redox enzymes, Protein Film Voltammetry (PFV) is a very fine tool. This technique, which we contribute to develop, was invented by Fraser Armstrong (now in Oxford) and applied to enzymes only 15 years ago.
In Marseilles, we use it to complement the results obtained from more traditional techniques. This collaboration with molecular biologists, spectroscopists, theoretical chemists and crystallographers makes it possible to probe structure-function relationships in “complex” (i.e. muticentered) redox enzymes.
Here is a tutorial, that uses hydrogenase as an example to explain what PFV is, and the reasons this is a useful technique for learning how enzymes work.
Hydrogenases.
Hydrogenases are complex metalloenzymes that catalyse the oxidation of di-hydrogen to protons and electrons, or the reductive formation of molecular hydrogen.
H2 <=> 2H+ + 2e–
The reaction occurs at an active site that is buried in the protein, and electrons are transferred to the redox partner via a chain of iron sulfur clusters.
Measuring (“assaying”) hydrogenase activity: the usual manner.
The principle of PFV is easily explained by comparing this technique with the more traditional solution assays of enzyme activity.
In homogeneous enzyme kinetics, the hydrogenase solution is mixed in a cuvette with H2 and a soluble, oxidized redox partner, which accepts the electrons resulting from H2 oxidation. The mediator changes color when it is reduced (e.g. Methyl viologen turns blue upon reduction) and so does the cuvette. The turnover frequency of the enzyme is determined by following the absorbance change of the solution.
This technique has several advantages.
- It is easy to implement
- It can measure the turnover rates even if it is small (just increase the concentration of enzyme until you detect a significant change in absorbance in a reasonable time).
But there are drawbacks:
- Measuring the activity typically takes several minutes, during which the activity must be constant.
- Furthermore, this measurement is not compatible with the presence of oxygen in the assay medium (O2 would directly and quickly oxidize the redox partner). It is unfortunate that a very hot topic is actually the study of hydrogenases that can work in air.
- Last, it is not easy to deal with gaseous substrates (H2) or inhibitors (O2, CO), unless you only need to keep their concentrations equal to that obtained in equilibrium with one atmosphere of either gas.
Assaying hydrogenase activity in electrochemistry
In mediated electrochemistry the consumption of the redox partner is detected as a current wave resulting from its electrochemical recycling on the electrode; only the mediator interacts with the electrode and the homogeneous catalytic process which occurs in the bulk of the electrochemical cell is fundamentally the same as in solution assays. Further reading
This is not what we do!
Using Protein Film Voltammetry

We are interested in a third configuration originally called « protein film voltammetry’’ (PFV), where the enzyme is adsorbed onto the electrode and electron transfer is direct. When the electrode potential is appropriate, electrons flow between the substrate and the electrode, via the active site of the enzyme, and the magnitude of the current is simply proportional to the turnover rate.
current = turnover frequency x electroactive coverage
Ideally, interfacial ET may be fast and the electrode rotated at high speed to avoid mass transport control, so that the current response directly reports on the intrinsic properties of the enzyme.
PFV has an important limitation and an obvious drawback:
- First, not all redox enzymes will stick to an electrode.
- Second, the electroactive coverage is often unknown, so that you cannot obtain the value of the turnover frequency (the specific activity) from the current…
This is a bad start.
However, the advantages of PFV make up for this:
The information is gained from relative changes in current.
In PFV, the absolute turnover frequency cannot always be measured, but any change in current informs about a relative change in activity. This may result from changing the electrode potential, or from changing the concentration of substrate or inhibitor, or from the enzyme (in)activating over time.
The electrode potential is a useful control parameter.
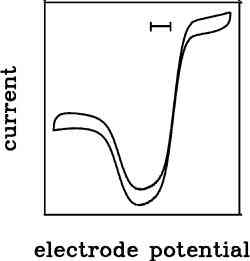
In homogeneous kinetics, the driving force (which depends on the concentration of redox partner) cannot be easily varied. In PFV the electrode potential is a natural control parameter.
By changing the electrode potential, the redox state of the enzyme changes, and the voltammogram (the plot of current against electrode potential) tells you about how the enzyme’s activity depends on its redox state. Clearly, this is an important information which you could not obtain from traditional assays, in homogeneous kinetics.
The profile of activity against potential may have a complex shape, which can be interpreted in relation to the catalytic mechanism. Further reading
High temporal resolution of the activity measurement.
In homogeneous kinetics, measuring the activity takes several minutes. In PFV the current can be sampled at high frequency (ten times per second or even faster). This is most useful when the activity of the enzymes is changing, for example the enzyme is being inhibited.
Hence we use chronoamperometry experiments (whereby the electrode potential is poised, and the current is measured as a function of time) to probe the kinetics of inhibition or (in)activation in great detail.
Easy manipulation of gaseous substrates and inhibitors.
For studying hydrogenases, you may need to measure the activity as a function of the concentration of substrate H2 or, inhibitor, e.g. CO, O2 or NO. But it may be difficult to maintain the partial pressure of either gas in a cuvette.
Since 2004, we have developed methods that make it very easy to use PFV to determine how the rate of turnover (or inhibition) depend on the concentrations of substrates (or inhibitors). It is always about measuring the activity as a function of time (in chronoamperometry) while the concentration of substrate/inhibitor is changing. And the concentration of dissolved gas is changed simply by flushing the electrochemical cell with another gas.
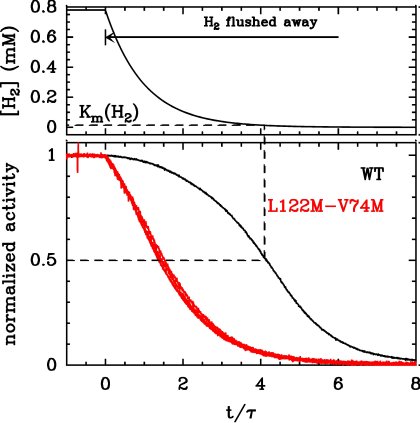
For example, to determine the Km for H2, measure the current under one atmosphere of H2 (by flushing the electrochemical cell with pure H2, the solubility of H2 is about 0.8mM), then from time t0, bubble argon in the solution. Argon flushes H2 away, the concentration of H2 decreases and so does the activity. The change in activity against time embeds the information about how the activity depends on the concentration of H2. Here is an example with WT D. fructosovorans NiFe hydrogenase, and a double mutant (L122M-V74M) whose Km is very large.
There is no need to independently measure the concentration of dissolved H2 because several independent control experiments have demonstrated that the change in concentration decays exactly exponentially. This makes data modelling very easy. Further reading
O2 does not interfere with the measurement of hydrogenase activity.
Solution assays of hydrogenase activity are not compatible with the presence of O2 because oxygen directly oxidizes the redox partner. In PFV, provided the electrode potential is sufficiently high, oxygen will not be reduced on the electrode and therefore it will not interfere with the activity measurement.
This is the reasoon PFV is useful for studying the aerobic inactivation mechanism of hydrogenases. For exmaple, here is how the activity of the FeFe hydrogenase from C. acetobutilycum evolves (bottom panel) when the enzyme is transiently exposed to O2 (the top panel is [O2] against time). From this sort of experiments, we’ve analyzed in detail the mecanism of inactivation of this enzyme.