The molecular process of electron bifurcation (EB) couples the endergonic 1-electron-reduction of a low-potential acceptor by two-electron redox compounds such as flavins or quinones to the exergonic 1-electron-transfer involving the other electron towards a relatively high-potential acceptor molecule. This mechanism represents one of the fundamental principles of free energy conversion in bioenergetics whose basic functional principles still aren’t fully understood (10.3389/fmicb.2018.01357).
Our group has been working on quinone-based electron bifurcation (QBEB) in bc1 and b6f complexes for more than 20 years and more recently also started to investigate this mechanism performed by flavin-containing enzymes, which is an emergent research subject with the first examples reported only a little over 10 years ago (10.1128/JB.01422-07; 10.1128/JB.01417-07).
The electron-bifurcating hydrogenase (HydABC) from Thermotoga maritima
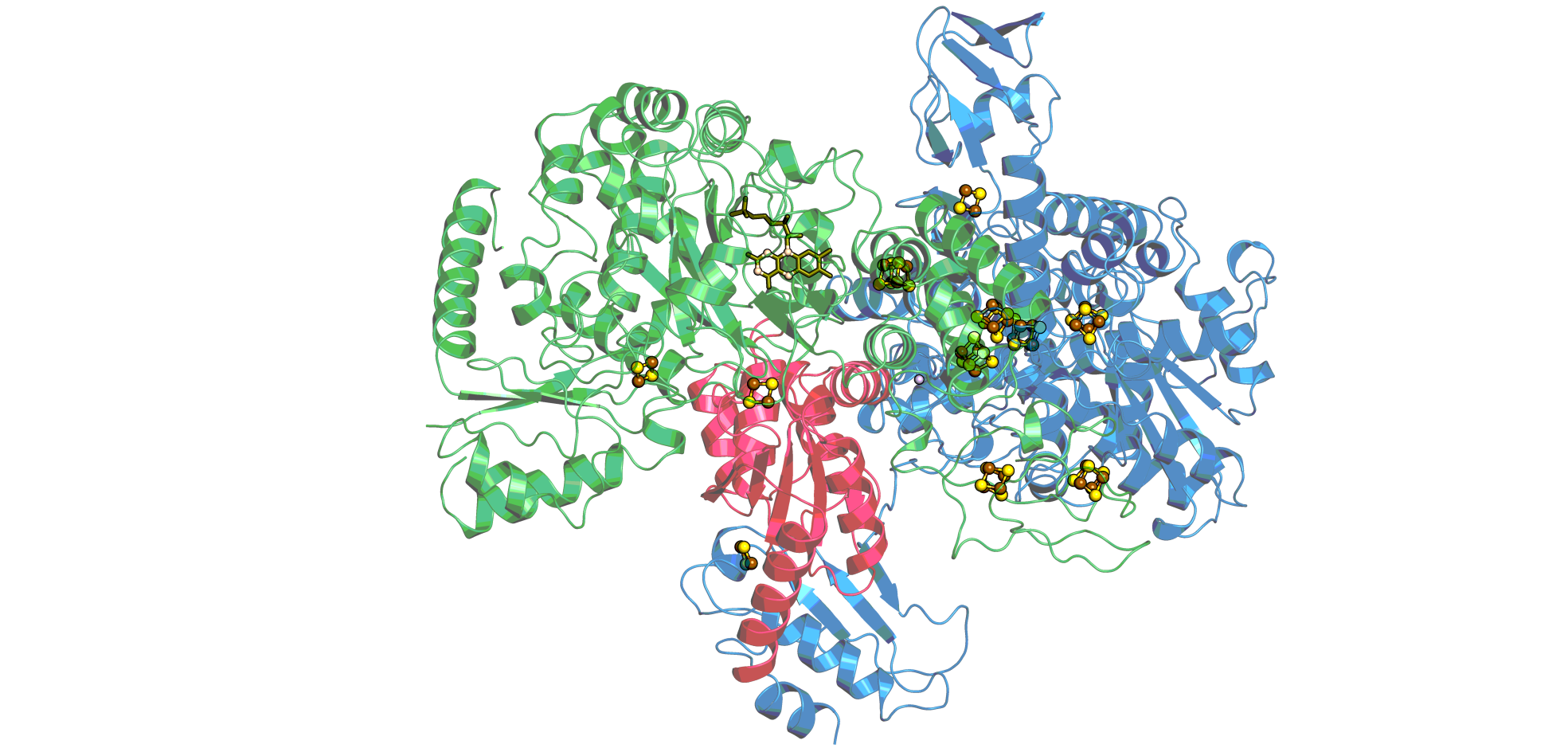
The electron bifurcating hydrogenase (HydABC) from Thermotoga maritimais a multi-centre protein complex containing flavin molecules together with a number of [2Fe-2S]- and [4Fe-4S]-clusters the redox properties of which are not yet well characterized. However, it is the first case of its enzyme family for which a high resolution cryoEM structure has been solved (10.7554/eLife.79361) and which therefore stands out by its strong potential for correlating structural and electrochemical parameters. It has been argued in the past that the ability to bifurcate electrons relies on the specific electrochemical properties of the redox centres forming the bifurcating unit, which, according to the recent structure of the Thermotoga enzyme, involves the flavin as well as one or two [2Fe-2S]- and one [4Fe-4S]-cluster.The collaboration project with the laboratory of James Birrell aims at determining the redox and pH-dependent properties of these centres.
Quinone-based electron bifurctaion occurs at the quinone-oxidizing (Qo) site of Rieske/cytochrome b complexes
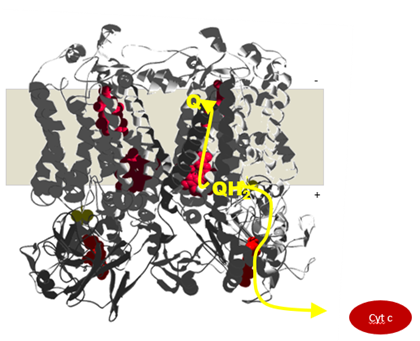
Electrons are transferred to a Rieske iron sulfur cluster (high potential chain) and to a low-potential b-heme (low potential chain). From the low-potential b-heme the electron is transferred across the membrane to another b-heme thereby building-up the proton motive force across the membrane

These reactions rely on a fine-tuned redox potentials of all co-factors:
- the redox midpoint potentials of the low-potential b-heme and the Rieske iron-sulfur cluster are higher and lower than the potential of the two electron transition of the quinone by the same amount.
- The redox potential difference between the two b-heme corresponds to the electrical part of the transmembrane potential
- The redox potential difference between the low potential b-heme and the Rieske iron-sulfur cluster is variable and depends on the species
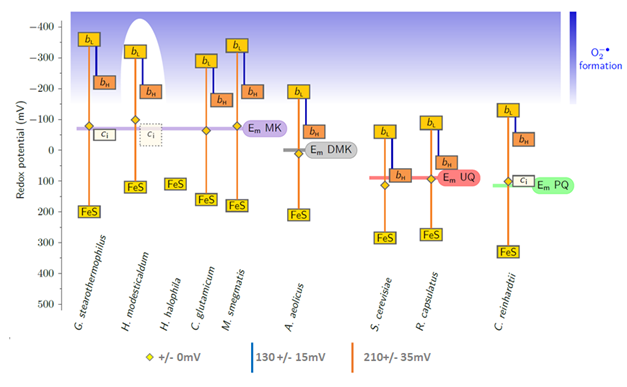
Note that the high redox midpoint potential of ubiquinones and plastoquinones, when compared to menaquinones allowed to take all the cofactros of the complex out of the redox range where reactions with oxygen occur. Only the Qo site semiquinone remains in the dangerous zone.
From low- to high-potential bioenergetic chains: Thermodynamic constraints of Q-cycle function
The modularity of proteins in various bioenergetic chains is of ongoing interest ever since the group was founded. By combining biochemical evidence and phylogenetical analysis we are able to trace back common ancestors and identify patterns that trace back to the protein make-up of the last universal common ancestor (LUCA).
The dyad of the Y-junction- and a flavin module unites diverse redox enzymes
![]() Our group seeks to understand the evolution of di-iron enzyme reaction centers as found in soluble methane monoxygenase (sMMO) or carbon monoxide dehydrogenase (CODH), and their analgous structural counterparts found in certain reactive naturally occuring minerals such as green rusts, mackinawite, and gregite. Insight into how abiotic geochemical pathways can replicate primative metabolic pathways and self-organize into structures condusive for the continuous production of organic precursors can help us understand how life began it’s inevitable journey to being.
Our group seeks to understand the evolution of di-iron enzyme reaction centers as found in soluble methane monoxygenase (sMMO) or carbon monoxide dehydrogenase (CODH), and their analgous structural counterparts found in certain reactive naturally occuring minerals such as green rusts, mackinawite, and gregite. Insight into how abiotic geochemical pathways can replicate primative metabolic pathways and self-organize into structures condusive for the continuous production of organic precursors can help us understand how life began it’s inevitable journey to being.
Our research:
Methanol on the rocks: Green rust transformation promotes the oxidation of methane
 Coming soon 🙂
Coming soon 🙂
We study selected prokaryotic energy conserving chains with the aim of contributing to a comprehensive picture of the diversity of these chains but also of elaborating common features and conserved principles.
The ultimate goal of this approach is to contribute to the elucidation of the origin(s) and evolutionary pathways of biological energy conversion.
All processes in living cells are energy-dependent. Apart from the comparatively inefficient fermentation pathway, this energy is invariably provided by the « Mitchellian » chemiosmotic mechanism, that is, by the conversion of a proton-motive-force (pmf) across a « bioenergetic membrane » into ATP-synthesis via the enzyme ATPase.
In the vast majority of cases, the pmf is produced by the coupling of electron transport to the vectorial translocation of positively charged ions (generally protons, H+) across the bioenergetic membrane (Figure 1). In a few archaeal species, the bioenergetic membrane potential is alternatively built up directly by light-driven ion-translocation performed by the enzyme Bacteriorhodopsin.

Life thus mainly draws its energy from collapsing electrochemical disequilibria of redox-active substrates. Such disequilibria may be provided by geo-/biochemical processes in the environment or induced by life itself via photochemical mechanisms (i.e. chlorophyll-based photosynthesis).
In eukaryotes, the corresponding electron transfer chains are almost invariably aerobic respiration (in mitochondria) or oxygenic photosynthesis (in chloroplasts). Prokaryotes, by contrast, feature a bewildering diversity of bioenergetic electron transfer chains and are probably able to use all bio-available redox compounds as substrates for energy conversion, either as reductants or oxidants. Figure 2 represents a few selected examples of this -probably far from exhaustively known- diversity of prokaryotic electron transfer chains.
BIP09 studies selected prokaryotic energy conserving chains with the aim of contributing to a comprehensive picture of the diversity of these chains but also of elaborating common features and conserved principles. The ultimate goal of this approach is to contribute to the elucidation of the origin(s) and evolutionary pathways of biological energy conversion